Why are some people unable to tolerate the TTFD form of thiamine? Did you know that TTFD temporarily depletes glutathione?
In this short series, we will be examining some of the potential reasons why certain individuals experience negative reactions and side effects from TTFD supplementation.
First of all, it is essential to understand the basics behind TTFDs molecular configuration and how it is processed by cells.
TTFD contains thiamine, but is NOT the SAME as thiamine. Simply put, the primary difference is an extra chemical group called a mercaptan group. This group is like what is also found in allicin, which is a compound found in garlic.
The mercaptan group is connected to the thiamine molecule via a special sulfur-sulfur bond called a disulphide bond. The unique chemical group is responsible for TTFD’s ability to traverse membranes in the body without the need for a transport system.
TTFD, with its special mercaptan group, is mostly absorbed whole as TTFD in the gastrointestinal tract. As it travels through the blood, it can enter the brain and many other organs. One of the main sites of absorption is actually in the red blood cells.
Upon penetration of the red blood cell membrane, TTFD must first be PROCESSED or “broken apart” before it can release the thiamine contained within its chemical structure. The ancillary mercaptan sulfur group must then be utilized and/or detoxified through alternative pathways.
I believe that main issues with tolerability are: 1. Errors in this processing or 2. Compromised detoxification of the sulfur groups.
HOW TTFD IS PROCESSED IN CELLS
For TTFD to “release” its thiamine, its disulfide bond must gain electrons from another donor molecule. In chemical terms, this process is referred to as chemical reduction. Once this reduction occurs, free thiamine is “released”.
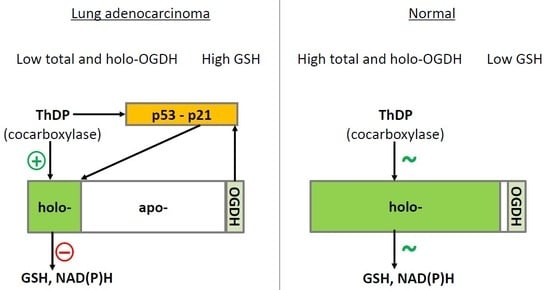
The molecule which has been shown to do this most effectively is GLUTATHIONE. Glutathione is the cell’s primary antioxidant. As an antioxidant, it can be found in its “reduced” form with an extra electron that can be donated, or its “oxidized” form after it has donated its electron. The process is as follows: Reduced glutathione (GSH) donates an electron, and so goes on to form oxidized glutathione (GSSG). GSSG is then recycled back to GSH through gaining electrons via the enzyme glutathione reductase (vitamin B2 dependent and NADPH).
In the context of TTFD – GSH in red blood cells chemically reduces TTFD via a process called “disulfide exchange” (using glutaredoxin) (1). Reduced glutathione becomes oxidized glutathione, TTFD “releases” thiamine, producing free thiamine inside the cell and an extra TFD group left over.
So in simple terms, to obtain thiamine from TTFD, you inevitably use up glutathione in the form of GSH. That’s right. The initial phase of processing TTFD requires that cells have enough reduced glutathione. Furthermore, the more GSH you have – the faster the rate of this reaction.
I recently corresponded with one individual who only gained tolerance of TTFD after supplementing with 200mcg of selenium in the form of sodium selenite. Selenium supplementation in different forms has been shown to increase red blood cell GSH levels by up to 35% (2). This is thought to occur due to selenium’s ability increase glutathione synthesis through upregulating the enzyme gamma-glutamylcysteine synthetase (3). I suspect that poor glutathione status might be one of the reasons for benefit from selenium.
Having enough glutathione is clearly very important, but recycling it is also essential to maintain a pool of glutathione in its reduced form. Unfortunately, TTFD places a burden on this system. This was demonstrated in one old study from Japan which showed that TTFD administration rapidly lowered GSH (4). However, in that same experiment GSH levels were restored within 5-10 minutes. This was accomplished by the vitamin B2 (as FAD)-dependent enzyme glutathione reductase, which donates electrons to GSSG with the reducing power of NADPH to recycle it back to GSH.
These are key points which might help us to understand why some people do not benefit from TTFD. First, cells need enough GSH to cleave thiamine. Second, cells also need to be able to recycle the glutathione which has become oxidized.
Immediately, we see two potential issues that could arise when someone supplements with TTFD.
First: In someone who has poor glutathione (GSH) status, they might theoretically be less able to generate thiamine from TTFD. There are many reasons why someone may have poor glutathione status.
- Low precursors (cysteine, glutamate, glycine)
- Chronic oxidative burden and/or inflammation
- Other nutrient deficiencies necessary to produce glutathione (such as B6 or selenium)
Some basic ways to measure glutathione status include: Whole blood glutathione, gamma-glutamyl-transpeptidase, pyroglutamic acid on an OAT
Second: Someone may have enough resources to make glutathione, but if they cannot RECYCLE it through glutathione reductase, then taking a substance which depletes their GSH further (like TTFD) might not be a good idea.
A total/functional riboflavin deficiency is the probably the main culprit when looking at poor glutathione reductase activity. The glutathione reductase enzyme also requires adequate NADPH to drive the enzymatic reaction. NADPH is derived from niacin (vitamin B3) but is also generated in the pentose phosphate pathway which, ironically, also requires thiamine. Restoring NADPH levels through supplementing with ordinary thiamine and supporting the pentose phosphate pathway via other means might be advised BEFORE starting with TTFD.
In the context of poor enzyme activity, without the reducing powder to drive GSSG back to GSH, the oxidized form of glutathione can theoretically drift towards the path of generating a free radical called the glutathione radical (5). This alone could further contributes to oxidative stress and cell damage.
How to know if RBC glutathione reductase activity is sufficient? This can be tested and is one of the markers for riboflavin status. Some other ways to assess riboflavin status include glutaric acid, whole blood B2, adipic, suberic, ethylmalonic acids, and urinary succinic acid can also be indicative.
Interestingly, here is one of the links between B1 and B2. Older research in Japan showed that TTFD supplementation could lead to a secondary B2 deficiency through increased urinary excretion (6). The increase need for glutathione reductase could at least also contribute to this effect. When taking TTFD, it has downstream effects on other nutrients. Hence, these supporting nutrients should probably also be taken in conjunction with high-dose supplementation.
To summarise, the initial cellular processing of TTFD requires adequate levels of reduced glutathione. Glutathione becomes oxidized, and so TTFD has can have a depleting effect on GSH and increase the requirement for recycling. If there is insufficient active B2 (as FAD) or NADPH levels, glutathione is not likely to be recycled sufficiently and may lead to GSSG radical formation.
It is therefore possible that the glutathione-depleting effect of TTFD could be responsible for some of the side effects associated with supplementation. This is probably most applicable in individuals with poor glutathione recycling and underlying oxidative stress.
Therefore, nutrient therapy which may support this initial phase of TTFD metabolism include:
- Selenium (improve GSH levels)
- Riboflavin (improve GSSG-GSH recycling)
- Niacin (increase NADPH)
- Ordinary thiamine (increase NAPH via PPP)
- NAC, glycine and/or glutathione TAKEN AWAY FROM TTFD (improve GSH status)
In the next piece, we will examine some of the following steps in the breakdown of TTFD with a focus on the nutrient cofactors and biochemical processes necessary for adequate clearance and detoxification of the mercaptan group. These include methylation, Phase I biotransformation, and sulfoxidation.
 www.mdpi.com
www.mdpi.com







") . Whichever brand one chooses, it provides the same raw ingredient. The one is a lower dose but is cheaper to buy, whereas the other is higher dose and more expensive per bottle.
. Whichever brand one chooses, it provides the same raw ingredient. The one is a lower dose but is cheaper to buy, whereas the other is higher dose and more expensive per bottle.


")

